
TÜRKİYE’DE TIBBİ CİHAZ ÜRETİCİSİYİM, UKRAYNA’YA İHRACAT YAPABİLMEM İÇİN GEREKLİLİKLER NELERDİR ?

Türkiye, tıbbi cihaz üretiminde bölgesel bir güç haline gelirken, Türk üreticiler için en stratejik pazarlardan biri de Ukrayna’dır. Ancak, Avrupa Birliği yönetmelikleriyle (MDR/MDD) benzerlik gösterse de, Ukrayna’nın kendine özgü prosedürleri bulunmaktadır. Özellikle Sınıf 1 (Class I) tıbbi cihaz üreticileri için süreç daha hızlı ilerlese de, Teknik Düzenleme No. 753 (Technical Regulation No. 753) gerekliliklerini eksiksiz yerine getirmek hayati önem taşır. https://moz.gov.ua/
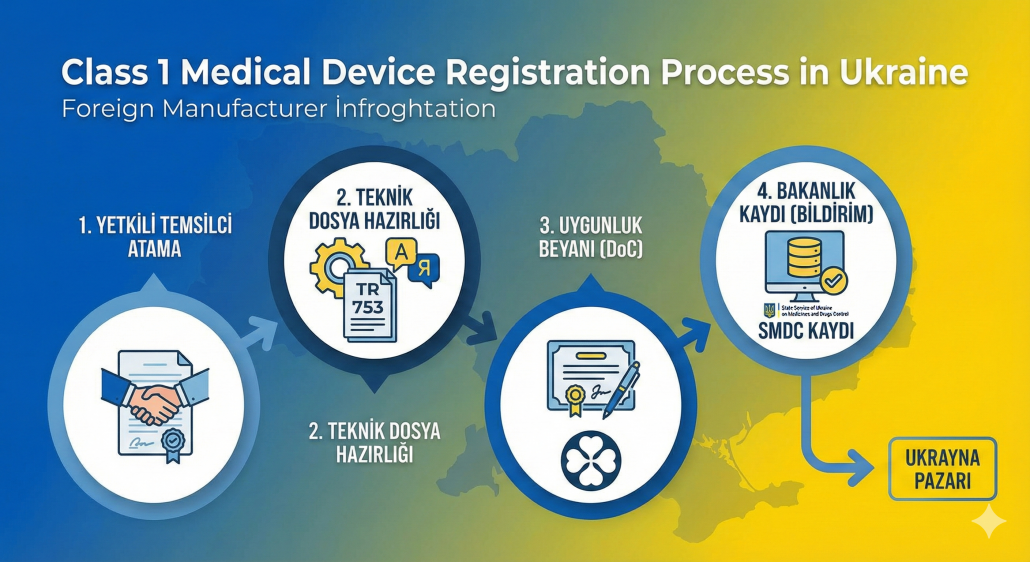
Bu yazımızda, Türkiye’de yerleşik bir üreticinin Sınıf 1 (steril olmayan, ölçüm yapmayan) cihazlarını Ukrayna Sağlık Bakanlığı sistemine nasıl kaydedeceğini adım adım inceliyoruz.
1. Temel Kural: Yerel Bir “Yetkili Temsilci” Şart
Ukrayna mevzuatına göre, ülkede yerleşik olmayan bir üretici (örneğin Türkiye’deki firmanız), ürünlerini doğrudan piyasaya süremez. Mevzuat gereği, Ukrayna’da yerleşik resmi bir Yetkili Temsilci (Authorized Representative) atamak zorundasınız.
Bu temsilci:
- Sizin adınıza Ukrayna makamlarıyla (SMDC) iletişim kurar.
- Teknik dosyanızı ve uygunluk beyanınızı 5 yıl (veya raf ömrü süresince) saklar.
- Ürün etiketlerinde adı ve adresi yer alır.
Önemli İpucu: Yetkili temsilci ile yapacağınız sözleşme ve vekaletname (Power of Attorney), noter onaylı ve Apostil şerhli olmalıdır.
Sınıf 1 Cihazlar İçin “Uygunluk Beyan” (Self-Declaration) Prosedürü
Sınıf 1 tıbbi cihazlar (örneğin; basit cerrahi aletler, yürüteçler, sargı bezleri vb.), en düşük risk grubunda yer aldığı için bir “Onaylanmış Kuruluş” (Notified Body) denetimine tabi değildir. Bunun yerine Öz Beyan prosedürü uygulanır.
Süreç şu şekilde işler:
- Teknik Dosya Hazırlığı: Ürününüzün tasarımını, üretim sürecini ve risk analizlerini içeren dosya hazırlanır.
- Kontrol Listesi (Ek-1): Teknik Düzenleme No. 753 Ek-1’de yer alan “Temel Gereklilikler” listesi doldurulur.
- Uygunluk Beyanı (Declaration of Conformity): Üretici olarak, ürününüzün Ukrayna mevzuatına uygun olduğunu beyan ettiğiniz resmi belgedir.
Etiketleme ve Dil Zorunluluğu: Ukraynaca Olmazsa Olmaz
İhracatçıların en sık yaptığı hata, İngilizce etiketlerin yeterli olacağını düşünmeleridir. Ukrayna’da satılacak her ürünün etiketi ve Kullanma Kılavuzu (IFU) Ukraynaca olmak zorundadır.
Etikette bulunması gerekenler:
- Üreticinin adı ve adresi.
- Ukrayna Yetkili Temsilcisinin adı ve adresi.
- Ulusal Uygunluk İşareti (Yonca İşareti): TR 753’e uygunluğu gösteren, ucu açık bir üçgeni andıran yonca sembolü (National Mark of Conformity).
Ukrayna Bakanlık Kaydı Nasıl Yapılır?
Sınıf 1 ürünler için süreç, bir “ruhsatlandırma”dan ziyade “Bildirim” (Notification) usulüdür.
- Hazırladığınız Uygunluk Beyanı ve Vekaletname, Ukrayna’daki temsilcinize gönderilir.
- Temsilci, bu belgelerle birlikte Ukrayna İlaç ve Uyuşturucu Kontrolü Devlet Hizmeti’ne (SMDC) başvurur.
- Yetkili kişiler veritabanına kayıt yapılır.
- Bu kayıt tamamlandığında, ürünleriniz Ukrayna gümrüklerinden sorunsuz geçerek piyasaya sürülebilir.
Sonuç: Ukrayna, Türk tıbbi cihaz üreticileri için büyük fırsatlar barındırıyor. Sınıf 1 cihazlarda süreç, doğru partnerler ve eksiksiz dokümantasyon ile oldukça hızlı tamamlanabilir. Mevzuata tam uyum, sadece gümrükten geçişi değil, pazardaki kalıcılığınızı da sağlar.

Ukrayna Bakanlar Kurulu’nun
11 Haziran 2008 tarihli ve 536 sayılı Kararı ile ONAYLANMIŞTIRhttps://zakon.rada.gov.ua/laws/show/753-2013-%D0%BF#Text
Tıbbi Cihazlara İlişkin Teknik Düzenlemeler
Genel kısım
1. Bu Teknik Yönetmelik, tıbbi cihazlara ilişkin genel şartları, bunların güvenliğini ve bu şartlara uygunluğun teyit edilmesine ilişkin prosedürleri tanımlamaktadır.
2. Teknik Yönetmelikte aşağıdaki terimler aşağıdaki anlamlarda kullanılmıştır:
1) devreye alma – tıbbi cihazın ilk amaçlanan kullanımına hazır olması;
2) piyasaya arz – klinik araştırmalara yönelik cihazlar hariç olmak üzere, bir tıbbi cihazın, Ukrayna pazarında amaçlanan amacı doğrultusunda dağıtım ve/veya kullanım amacıyla ilk kez ortaya çıkması, bu cihazın yeni veya tamamen yenilenmiş olup olmadığına bakılmaksızın;
3) Tıbbi cihazlar – invaziv olanlar ve insan vücudunda temel tedavi amacını gerçekleştirmek için değil, bu amacın gerçekleştirilmesinde farmakolojik, immünobiyolojik veya metabolik ajanların işlevlerini desteklemek için tasarlanmış olanlar da dahil olmak üzere her türlü alet, cihaz, cihaz, ekipman, implant, malzeme veya diğer ürünler ile üretici tarafından sağlandığı şekilde, uygun şekilde kullanılmaları için gerekli yazılım araçları da dahil olmak üzere, hem ayrı ayrı hem de birbirleriyle kombinasyon halinde kullanılan ürünler, aşağıdakileri sağlamak için:
bir hastanın hastalığı, yaralanması, sakatlığı halinde veya bir organ eksikliği veya fiziksel kusurunun tazmini için hastanın durumunun önlenmesi, teşhisi, tedavisi, izlenmesi veya hafifletilmesi;
organların, dokuların veya fizyolojik süreçlerin yapısının (anatomisinin) araştırılması, değiştirilmesi veya değiştirilmesi;
Döllenme sürecinin kontrolü.
Tıbbi cihazlarla birlikte verilen ve diğer harici (ek) ekipmanlarla birleştirilmesi amaçlanan her türlü ekipman, söz konusu tıbbi cihazların ayrılmaz bir parçası olarak kabul edilir;
4) Özel olarak üretilen tıbbi cihazlar: Uygun niteliklere sahip bir doktorun reçetesi doğrultusunda özel olarak üretilen, belirli özelliklere ve tasarıma sahip ve yalnızca belirli bir hasta için tasarlanmış tıbbi cihazlardır.
Belirtilen reçete, uygun niteliklere sahip başka bir kişi tarafından da düzenlenebilir.
Nitelikli bir hekimin veya herhangi bir profesyonel kullanıcının özel gereksinimlerini karşılamak için değişiklik gerektiren seri üretim tıbbi cihazlar, özel yapım ürünler değildir;
5) Klinik araştırmalara yönelik tıbbi cihazlar – Klinik ortamlarda araştırma yapma sürecinde uygun niteliklere sahip bir hekim tarafından kullanılması amaçlanan tıbbi cihazlar.
Klinik araştırmanın yürütülmesi sırasında, niteliklerine göre böyle bir araştırmayı yürütme hakkına sahip olan herhangi bir diğer kişi, uygun niteliklere sahip bir hekimle eşdeğer tutulur;
6) amaçlanan kullanım – tıbbi cihazların, üretici tarafından etiketleme, kullanım talimatları ve/veya reklam materyallerinde belirtilen verilere uygun olarak kullanılması;
7) Aksesuarlar – Tıbbi cihaz olmayan, ancak üretici tarafından tıbbi cihazlarla birlikte kullanım amacına uygun olarak kullanılmak üzere özel olarak tasarlanmış ürünler.
Diğer terimler, Ukrayna Yasalarında verilen anlamlarda kullanılmaktadır: “Uygunluk Onayı Hakkında”, “Standardizasyon Hakkında”, “Uygunluk Değerlendirme Kuruluşlarının Akreditasyonu Hakkında”, “Standartlar, Teknik Düzenlemeler ve Uygunluk Değerlendirme Prosedürleri Hakkında” ve “Tıbbi Ürünler Hakkında”.
3. Bu Teknik Yönetmelikte belirlenen şartlar aşağıdakiler için zorunludur:
tıbbi cihaz ve aksesuar üreticileri;
yetkili üreticiler – Ukrayna’da ikamet edenler (bundan böyle yetkili temsilci olarak anılacaktır);
Üretici veya yetkili temsilcisi Ukrayna topraklarında faaliyet göstermiyorsa tıbbi cihazları piyasaya arz eden veya bunları işletmeye alan kişiler (bundan böyle tıbbi cihazları piyasaya arz eden veya bunları işletmeye alan kişi olarak anılacaktır);
tıbbi cihazlar ve aksesuarlarının güvenliğinin teknik düzenlemesi ve denetimi işlevleriyle görevli merkezi yürütme makamları (bundan sonra merkezi yürütme makamları olarak anılacaktır);
Tıbbi cihaz ve aksesuarların uygunluğunu değerlendirmek üzere yetkilendirilmiş kuruluşlar, bu kuruluşlara ilişkin gereklilikler Ukrayna Bakanlar Kurulu’nun 24 Ocak 2007 tarihli ve 59 sayılı ” Ürünlerin, Proseslerin ve Hizmetlerin Teknik Düzenlemelerin Gereksinimlerine Uygunluğunu Değerlendirmek İçin Kuruluşların Belirlenmesine İlişkin Prosedürün Uygulanmasına İlişkin Prosedürün Onaylanması Hakkında” (Ukrayna Resmi Gazetesi, 2007, No. 6, s. 223) Kararı ile belirlenmiştir (bundan böyle yetkilendirilmiş kuruluşlar olarak anılacaktır).
4. İnsan vücuduna bir tıbbi ürünün uygulanması için tasarlanmış bir tıbbi cihaz, tıbbi ürüne ilişkin gereklilikler uyarınca bu Teknik Yönetmeliklere ve Ukrayna “Tıbbi Ürünler Hakkındaki” Kanuna tabidir.
Bir tıbbi ürün ve bir tıbbi cihaz tek bir bütün teşkil ediyorsa ve tıbbi cihaz tekrar kullanıma uygun değilse, söz konusu ürün Ukrayna “Tıbbi Ürünler Hakkında” Kanunu’na tabidir. Bu Teknik Yönetmelik ile belirlenen gereklilikler, yalnızca tıbbi cihazın güvenliği ve etkinliği ile ilgili özellikleri için geçerlidir.
Bir tıbbi cihaz, insan vücudu üzerinde yardımcı etki gösterebilen bir aracı bütünleyici bir parça olarak içeriyorsa ve kullanıldığında tıbbi ürün olarak değerlendiriliyorsa, söz konusu tıbbi cihaz bu Teknik Yönetmelik hükümlerine uygun olmalıdır.
5. Tıbbi cihazların elektromanyetik uyumluluğu
DSTU IEC 60601-1-2-2001 “Tıbbi Elektrikli Ekipmanlar” standardı ile düzenlenmektedir.
Elektromanyetik uyumluluğun doğrulanmasına ilişkin bu Teknik Yönetmelik tıbbi cihazlara uygulanmaz.
6. Tıbbi cihazlar, kullanım potansiyel riskine bağlı olarak I, IIa, IIb ve III sınıflarına ayrılır. Tıbbi cihazların belirli bir sınıfa atanması, insan vücudunun hassasiyetine, bu cihazların geliştirilmesi ve üretimiyle ilişkili potansiyel riskler dikkate alınarak yapılır ve DSTU 4388:2005 “Tıbbi cihazlar. Kullanım potansiyel riskine bağlı sınıflandırma. Genel gereklilikler” standardında tanımlanan kriterler ve sınıflandırma kuralları kullanılarak gerçekleştirilir.
Tıbbi cihazın sınıfının belirlenmesi konusunda üretici ile yetkili kuruluş arasında uyuşmazlık çıkması halinde, konu Sağlık Bakanlığı tarafından belirlenen usule göre çözümlenir.
7. Bu Teknik Yönetmeliğe tabi tıbbi cihazların piyasaya arzı ve çalıştırılması, hastaların, kullanıcıların ve diğer kişilerin yaşam ve sağlığı için bir tehdit oluşturmaması, usulüne uygun olarak kurulması, bakımının yapılması ve amacına uygun olarak kullanılması kaydıyla mümkündür.
8. Tıbbi cihazları piyasaya arz eden veya hizmete sunan üretici veya onun yetkilendirdiği kişi, bu Teknik Yönetmelikte öngörülen tüm uygunluk değerlendirme işlemlerini mevzuata uygun olarak gerçekleştirmekle yükümlüdür.
Bu tür gereklilikler, bitmiş tıbbi cihazları monte eden, paketleyen, yeniden işleyen ve/veya etiketleyen tüzel kişiler ve gerçek kişiler için de geçerlidir.
Bu şartlar, üretici olmayan, ancak belirli bir hasta için amaçlanan kullanım amacıyla piyasaya arz edilmiş tıbbi cihazları monte eden veya çalıştıran kişiler için geçerli değildir.
9. Ulusal standartların gereklerini karşılayan tıbbi cihazlar, ürünün bu Teknik Yönetmelik gereklerine uygun olduğunun kanıtıdır.
Devlet Tüketiciyi Koruma ve Standardizasyon Komitesi tarafından sunulan ulusal standartlar listesi, cerrahi dikişler ve tıbbi ürünler ile bu tür tıbbi ürünleri içeren tıbbi cihazlarda kullanılan malzemeler arasındaki etkileşimlere ilişkin Avrupa Farmakopesi monografilerini içermektedir.
10. Bu Teknik Yönetmelikte belirlenen usullere uygun olarak uygunluk değerlendirmesinden geçen tıbbi cihazlar, dolaşıma sokulmadan veya hizmete sokulmadan önce, Ukrayna Bakanlar Kurulu’nun 29 Kasım 2001 tarihli ve 1599 sayılı “Ulusal Uygunluk İşaretinin Tanımı ve Uygulanmasına İlişkin Kuralların Onaylanması Hakkında” Kararı (Ukrayna Resmi Gazetesi, 2001, No. 49, s. 2188) uyarınca ulusal uygunluk işareti ile işaretlenmelidir.
11. Tıbbi cihazlarda ulusal uygunluk işaretinin bulunması, işaretlemeyi yapan veya işaretlemeden sorumlu olan gerçek veya tüzel kişinin, tıbbi cihazların bu Teknik Yönetmelikte yer alan cihazlara ilişkin gerekliliklere uygunluğunu kontrol edip onayladığı ve uygun uygunluk değerlendirme prosedürlerinin tamamlandığını ifade eder.
12. Tıbbi cihazlar, ulusal uygunluk işaretinin iliştirilmesini öngören diğer teknik düzenlemelere tabi ise, tıbbi cihazlar bu teknik düzenlemelerin gerekliliklerine de uygun olmalıdır. İşaretin iliştirilmesinin koşulu, tıbbi cihazların bu teknik düzenlemelerin tamamına uygun olmasıdır.
Bir veya daha fazla teknik düzenleme, imalatçının uygunluk teyidi yöntemini seçmesini öngörüyorsa, ulusal uygunluk işaretinin iliştirilmesi, yalnızca imalatçı tarafından uygulanan teknik düzenlemelere uygunluğu gösterir. Bu durumda, tıbbi cihazlara eşlik eden belgelerde, duyurularda veya talimatlarda uygulanan teknik düzenlemelere atıflar yapılır.
13. Ulusal uygunluk işaretinin bu Teknik Yönetmelik hükümlerine aykırı olarak kullanıldığı tespit edilirse, imalatçı veya yetkili temsilcisi ya da tıbbi cihazları piyasaya arz eden veya hizmete sunan kişi, ihlali ortadan kaldırmak, tıbbi cihazları bu Teknik Yönetmelik hükümlerine uygun hale getirmek ve bu uygunluğu belirlenen usule göre teyit etmek için gerekli tedbirleri almak zorundadır.
Mevzuata aykırılık halinde, merkezi yönetim organları, söz konusu tıbbi cihazların piyasaya arzını kısıtlamak veya yasaklamak ya da piyasadan çekmek için gerekli tedbirleri alır.
14. Özel olarak üretilen tıbbi cihazlar, bu Teknik Yönetmeliğin 54 üncü maddesinde belirtilen şartları karşılıyorsa, bu cihazlara ulusal uygunluk işareti iliştirilmez.
15. Hekimlerin klinik araştırma amaçlı tıbbi cihazları kullanmaları ve bu Teknik Yönetmeliğin 56 ncı maddesinde belirtilen şartlara uymaları halinde, bu cihazlara ulusal uygunluk işareti konulmaz.
16. Bu Teknik Yönetmelik hükümlerine uygun olarak uygunluk değerlendirme işlemlerinden geçmemiş tıbbi cihazların, imalatçı tarafından bu Teknik Yönetmelik hükümlerine uygun hale getirilene kadar piyasaya arz edilemeyeceği veya işletmeye alınamayacağı yönünde açık bir işaretle işaretlenmesi koşuluyla, fuarlarda, sergilerde ve gösterilerde sergilenmesine izin verilir.
17. Başvuru sonuçlarına göre Sağlık Bakanlığı , dolaşıma sokulan veya işletilen tıbbi cihazlara ilişkin olarak aşağıdaki hususlarda sürekli kayıt tutar ve bilgileri değerlendirir :
Ürünlerin özelliklerinde ve/veya niteliklerinde meydana gelen arızalar veya bozulmalar, etikette veya kullanım kılavuzunda yer alan bilgilerde herhangi bir tutarsızlık olması ve bu durumun hastaların, kullanıcıların ve diğer kişilerin ölümüne veya sağlıklarında ciddi bozulmalara yol açabilecek veya açmış olması;
Ürünlerin özelliklerinde ve/veya niteliklerinde meydana gelen teknik veya tıbbi nedenlerle meydana gelen değişiklikler, bu ürünlerin üreticisi tarafından sistematik olarak geri çağrılmasına yol açar.
Kayıt tutma ve analiz için hekimler ve sağlık kuruluşları belirtilen bilgileri Sağlık Bakanlığı’na, üreticilere veya bunların yetkili temsilcilerine sunarlar.
18. Tıbbi cihazların, usulüne uygun olarak monte edilmesi, bakımı ve kullanımı sırasında hastaların, kullanıcıların ve diğer kişilerin sağlığı ve güvenliği açısından risk oluşturabileceğinin tespit edilmesi halinde, bu cihazların piyasadan çekilmesi veya mevzuata uygun olarak piyasaya arzının veya kullanımının yasaklanması veya kısıtlanması için mümkün olan her türlü tedbir alınır.
Tıbbi cihazların güvenliğine ilişkin gereklilikler
19. Tıbbi cihazların kullanım amacı, hastaların, kullanıcıların ve diğer kişilerin sağlık ve güvenliği açısından risk oluşturmamalıdır.
Tıbbi cihazların kullanımıyla ilişkili olabilecek olası risklerin, hastaya sağlayacağı yararlı etkiyle kıyaslandığında kabul edilebilir düzeyde olduğu ve yaşam ve sağlığın yüksek düzeyde korunmasıyla birleştiği varsayılmaktadır.
20. Tıbbi cihazlar, üretici tarafından öngörülen performans özelliklerini karşılamalı ve üretici tarafından belirtilen işlevleri yerine getirmeye uygun olacak şekilde tasarlanmalı, üretilmeli ve paketlenmelidir.
21. Bir tıbbi cihaz, imalatçı tarafından öngörülen uygun kullanım veya depolama koşulları altında çalıştırıldığında, imalatçı tarafından belirlenen süre içerisinde, bu Teknik Yönetmeliğin 25-44 üncü maddelerinde belirtilen özelliklerini ve/veya niteliklerini değiştirebilir; ancak bu özellikler ve nitelikler, hastaların, kullanıcıların ve diğer kişilerin klinik durumunu, güvenliğini tehlikeye atacak ölçüde bozulmamalıdır.
22. Tıbbi cihazlar, üreticinin öngördüğü koşullar altında, nakliye, kullanım veya depolama sırasında özelliklerinin ve/veya niteliklerinin bozulmayacak şekilde tasarlanmalı, üretilmeli ve paketlenmelidir.
23. Tıbbi bir cihazın istenmeyen herhangi bir yan etkisi, amaçlanan etkiye kıyasla kabul edilebilir bir potansiyel risk oluşturmalıdır.
24. Üreticinin tıbbi cihazların tasarımı ve üretimi sırasında aldığı kararlar, güncel teknolojiyi dikkate alarak güvenlik gerekliliklerini karşılamalıdır.
Üretici karar alırken öncelikle aşağıdaki ilkelere göre hareket etmelidir:
Tıbbi cihazların kullanımından kaynaklanan potansiyel risklerin mümkün olduğunca ortadan kaldırılması veya azaltılması;
tıbbi cihazların kullanımından kaynaklanan ve ortadan kaldırılması mümkün olmayan potansiyel riskleri önlemek için sinyal cihazları da dahil olmak üzere uygun koruyucu önlemlerin alınması;
Uygun koruyucu önlemlerin alınamaması nedeniyle tıbbi cihazların kullanımında ortaya çıkabilecek potansiyel riskler konusunda kullanıcıları bilgilendirmek.
Tıbbi cihazların geliştirilmesi ve üretimi için gereklilikler
bu Teknik Yönetmeliğin 25-50 nci maddelerinde belirtilen özellik ve/veya nitelikleri taşıyacak şekilde tasarlanır, üretilir ve paketlenir .
Bu durumda özellikle şunlara dikkat edilmelidir:
malzemelerin seçimi, özellikle toksisiteleri ve gerekirse yanıcılıkları açısından;
Ürünün kullanım amacı dikkate alınarak, malzemelerin insan vücudunun dokuları, hücreleri ve sıvılarıyla uyumluluğu;
Ürünün amacını da göz önünde bulundurarak, tıbbi cihazlarla bulaşma riskini ve bunların taşınması, depolanması ve kullanımında görev alan kişiler ile hastalar üzerindeki etkiyi en aza indirmek;
insan dokusu üzerindeki etkileri, bu etkinin süresi ve sıklığı;
Uygun şekilde kullanıldığında veya işlemler sırasında temas ettikleri malzeme, madde ve gazlarla kullanım güvenliği;
Ekipmanlardan madde sızıntısından kaynaklanan risklerin en aza indirilmesi;
Ürünün tasarımı, malzemesi ve kullanılacağı ortam göz önünde bulundurularak, tehlikeli maddelerin kazara ürüne girme riskinin en aza indirilmesi;
hasta, kullanıcı ve diğer kişilerin enfeksiyon riskini ortadan kaldırmak veya en aza indirmek.
26. Tıbbi ürünlerin uygulanmasına yönelik cihazlar, bu tür tıbbi ürünlerin kullanımına ilişkin belirlenmiş gerekliliklere uygun olarak, bu tür tıbbi ürünlerle uyumluluğu dikkate alınarak tasarlanmalı ve üretilmeli ve özelliklerinin kullanım amaçlarına uygun olarak muhafaza edilmesi sağlanmalıdır.
27. Bir cihaz, tek başına kullanıldığında tıbbi ürün olarak değerlendirilebilecek ve insan vücudu üzerinde cihazın etkisini tamamlayacak bir etki yaratması amaçlanan bir maddeyi bütünleyici bir parça olarak içeriyorsa, söz konusu ürünün güvenliği, kalitesi ve etkililiği, cihazın kullanım amacı dikkate alınarak, belirlenmiş prosedüre uygun olarak doğrulanmalıdır.
28. Tıbbi cihazlarda kullanım amacına uygun olarak kullanılan hayvansal dokular, devlet veterinerlik ve sağlık kontrolünden geçmiş hayvanlardan elde edilir.
Devlet veterinerlik makamları bu tür hayvanların bölgesel kökenine ilişkin bilgileri saklarlar.
Hayvansal kökenli doku, hücre ve maddelerin işlenmesi, depolanması, test edilmesi ve elleçlenmesi azami korumayla gerçekleştirilmektedir. Özellikle, üretim sürecinde onaylı imha veya viral inaktivasyon yöntemleri kullanılarak virüslere ve diğer enfeksiyon vektörlerine karşı koruma sağlanmaktadır.
29. Steril olarak tedarik edilen tıbbi cihazlar, onaylı yöntemlere uygun olarak uygun koşullar altında tasarlanmalı, üretilmeli, sterilize edilmeli ve tek kullanımlık ambalajlarda ve/veya piyasaya arz edildiğinde sterilliğini garanti eden uygun prosedürlere uygun olarak paketlenmeli ve koruyucu ambalajı hasar görene veya açılana kadar uygun koşullar altında saklanmalı ve taşınmalıdır.
30. Steril olmayan tıbbi cihazlar için ambalaj sistemleri, bu tür cihazlar için uygun bir temizlik seviyesini sağlamalı, kullanımdan önce sterilizasyon yapılması planlandığında bakteriyel kontaminasyon riskini en aza indirmeli ve üretici tarafından belirtilen sterilizasyon yöntemi dikkate alınarak kullanıma uygun olmalıdır.
31. Ambalaj ve/veya etiketleme, hem steril hem de steril olmayan şekilde tedarik edilen aynı veya benzer tıbbi cihazlar arasında ayrım yapabilecek nitelikte olmalıdır.
Tıbbi cihazların tasarımı ve çevresel özellikleri
32. Tıbbi cihazların diğer cihaz veya ekipmanlarla birlikte kullanılması amaçlanıyorsa, bağlantı sistemi de dahil olmak üzere bu kombinasyon güvenli olmalı ve cihazların performansını etkilememelidir. Bu tür cihazların kullanımına ilişkin kısıtlamalar, etikette veya kullanım talimatlarında belirtilmelidir.
33. Tıbbi cihazlar aşağıdaki riskleri en aza indirecek şekilde tasarlanmalı ve üretilmelidir:
Hacim-basınç oranı, boyut ve ergonomik özellikler dahil olmak üzere fiziksel özelliklerle ilgili yaralanmalara neden olmak;
manyetik alanlar, elektriksel etkiler, elektrostatik deşarj, basınç, sıcaklık veya basınç ve ivmedeki değişiklikler gibi makul ölçüde beklenen dış koşullarla ilişkili;
araştırma veya tedavi amacıyla kullanılan diğer ürünlerle etkileşim;
kullanılan malzemelerin eskimesi veya herhangi bir ölçüm veya kontrol mekanizmasının doğruluğunun azalması nedeniyle bakım veya kalibrasyonun (implantasyon durumunda olduğu gibi) imkansızlığı ile ilişkilidir.
34. Tıbbi cihazlar, uygun şekilde kullanıldığında ve tek bir arıza durumunda yangın veya patlama riskini en aza indirecek şekilde tasarlanmalı ve üretilmelidir.
Özellikle yanıcı veya tutuşmaya neden olabilecek maddelere maruz kalması beklenen cihazlara dikkat edilmelidir.
Ölçüm fonksiyonlu tıbbi ürünler
35. Ölçme fonksiyonlu tıbbi cihazlar, kullanım amaçları dikkate alınarak, gerekli sınırlar içinde yeterli doğruluk ve kararlılığı sağlayacak şekilde tasarlanmalı ve üretilmelidir.
Ölçüm, kontrol ve gösterge terazisi, ürünün kullanım amacı göz önünde bulundurularak ergonomik prensiplere göre tasarlanmıştır.
Ölçüm fonksiyonu olan tıbbi cihazlar kullanılarak yapılan ölçümler SI birimleri cinsinden raporlanır.
Radyasyon fonksiyonlu tıbbi ürünler
36. Radyasyon fonksiyonlu tıbbi cihazlar, aşağıdakileri önleyecek şekilde tasarlanmalı ve üretilmelidir:
terapötik ve tanısal amaçlara ulaşmak için gerekli olan düzeylerle sınırlı olmaksızın hastaların, kullanıcıların ve diğer kişilerin maruz kalması;
hastalara, kullanıcılara ve diğer kişilere zararlı radyasyon yaymaktadır.
Tıbbi cihazlar, belirli bir tıbbi amaç için gerekli olan ve faydasının maruz kalma riskinden daha ağır bastığı düşünülen tehlikeli düzeyde radyasyon yaymayı amaçlıyorsa, kullanıcı bu radyasyonu kontrol edebilmelidir. Bu tür cihazlar, belirtilen parametrelerin tekrarlanabilirliğini ve toleranslarını sağlayacak şekilde tasarlanmalı ve üretilmelidir.
Tıbbi cihazların potansiyel olarak tehlikeli görünür ve/veya görünmez radyasyon üretmesi amaçlanıyorsa, bu tür radyasyona karşı görsel ve/veya işitsel uyarı araçlarıyla donatılması gerekir.
Radyasyon fonksiyonuna sahip tıbbi cihazların kullanım talimatlarında, radyasyonun türleri, hastaların, kullanıcıların ve diğer kişilerin korunma yolları ve uygunsuz kullanımının önlenmesine ilişkin ayrıntılı bilgiler yer almalıdır.
37. İyonlaştırıcı radyasyon fonksiyonlu tıbbi cihazlar, amaçlarına uygun olarak geliştirilir ve üretilir, radyasyonun niceliksel ve niteliksel özelliklerinin düzenlenmesi ve kontrolü sağlanır.
38. X-ışını tanısında kullanılmak üzere tasarlanmış iyonlaştırıcı radyasyon fonksiyonlu tıbbi cihazlar, amaçlanan tıbbi amaçlar için gerekli görüntü kalitesi ve/veya çıktı göstergelerinin elde edilmesini sağlayacak ve aynı zamanda hastalar, kullanıcılar ve diğer kişiler için maruz kalma riskini en aza indirecek şekilde tasarlanmalı ve üretilmelidir.
39. X-ışını tedavisi için tasarlanan iyonlaştırıcı radyasyon fonksiyonlu tıbbi cihazlar, optimum radyasyon dozunun, ışın tipinin ve gücünün ve gerektiğinde radyasyon yoğunluğunun güvenilir bir şekilde kontrol edilmesini ve düzenlenmesini sağlayacak şekilde tasarlanmalı ve üretilmelidir.
Güç kaynağına bağlı veya güç kaynağıyla donatılmış tıbbi cihazlar için gereklilikler
40. Elektronik yazılım sistemlerine sahip tıbbi cihazlar, ürünün kullanım amacına uygun olarak tekrarlanabilirliğini, güvenilirliğini ve etkinliğini sağlayacak şekilde tasarlanmalı ve üretilmelidir. Bir sistem arızası durumunda, ortaya çıkan riski ortadan kaldırmak veya azaltmak için uygun önlemler alınmalıdır.
Hastaların güvenliğinin kullanımı sırasında dahili bir güç kaynağına bağlı olduğu tıbbi cihazlar, bu kaynağın durumunu belirleme olanağı sağlayan araçlarla donatılmıştır.
Hastaların güvenliğinin harici bir güç kaynağına bağlı olduğu tıbbi cihazlar, elektrik kesintisi durumunda uyarı veren bir alarm sistemiyle donatılmıştır.
Hastanın bir veya birden fazla klinik parametresini izlemek amacıyla tasarlanan tıbbi cihazlar, hastanın, kullanıcının ve diğer kişilerin ölümüne veya sağlıklarında ciddi bozulmaya yol açabilecek durumlar konusunda kullanıcıyı uyaran bir alarm sistemiyle donatılmıştır.
41. Bir güç kaynağına bağlı olan veya böyle bir güç kaynağıyla donatılmış tıbbi cihazlar, aşağıdaki önlemler alınarak tasarlanmalı ve üretilmelidir:
Doğru kullanıldığında diğer ürün veya ekipmanların çalışmasını etkileyebilecek elektromanyetik alan riskinin önlenmesi;
Hem uygun kullanım sırasında hem de tek bir ihlal durumunda, doğru şekilde monte edilmeleri koşuluyla elektrik çarpması riskini mümkün olduğunca ortadan kaldırmak;
hastaları, kullanıcıları ve diğer kişileri cihazın ve hareketli parçaların sağlamlığı ve dengesiyle ilişkili tehlikelerden korumak;
Ürünlerin yarattığı titreşimlerden kaynaklanan tehlikenin, özellikle titreşimin kaynağında sınırlandırılmasına yönelik tedbirler dikkate alınarak mümkün olan en düşük seviyeye indirilmesi, ancak çalışma karakteristiklerinin doğasında bulunan titreşimler hariç;
Gürültünün çalışma karakteristiklerinin bir parçası olması durumunda, özellikle kaynağında gürültüyü sınırlama araçları dikkate alınarak, oluşan gürültüden kaynaklanan tehlikenin mümkün olan en düşük seviyeye indirilmesi.
Kullanıcının temas ettiği elektrik, gaz veya hidrolik ve pnömatik enerji kaynağına sahip tıbbi cihazın terminalleri ve konnektörleri, tüm olası riskleri en aza indirecek şekilde tasarlanmalı ve üretilmelidir.
42. Tıbbi cihazların erişilebilir parçaları (ısı veya belirli sıcaklıklara kadar ısı sağlamak amacıyla kullanılan parçalar veya alanlar hariç) ve bunların etrafındaki alan, uygun şekilde kullanıldığında potansiyel olarak tehlikeli sıcaklıklara kadar ısınmamalıdır.
43. Hastalara enerji veya madde iletmek amacıyla tasarlanan tıbbi cihazlar, iletim yoğunluğunun hastanın, kullanıcının ve diğer kişilerin güvenliği için yüksek doğrulukta oluşturulmasını ve sürdürülmesini sağlayacak şekilde tasarlanmalı ve üretilmelidir.
44. Tehlike yaratabilecek enerji beslemesinin yoğunluğundaki tutarsızlığı önlemek ve/veya göstermek için araçlarla donatılmış, mümkün olduğunca tehlikeli seviyelerde enerji ve/veya madde kaynaklarından kazara beslemeyi önlemek için uygun araçlar içeren, kontrol ve gösterge işlevlerinin açık ve anlaşılır bir şekilde gösterildiği tıbbi cihazlar.
Tıbbi cihazın çalışması için gerekli olan, güncel ve/veya ayarlanabilir parametreleri gösteren talimatlar veya işaretler bulunuyorsa, bu bilgiler kullanıcı ve hasta tarafından anlaşılabilir olmalıdır.
Üretici tarafından sağlanan bilgiler
45. Her tıbbi cihaz, kullanıcıların eğitim ve yeterlilikleri ile üretici firmanın kimliği de dikkate alınarak, güvenli kullanımı için gerekli bilgilerle birlikte sunulmalıdır.
Üreticinin talebi üzerine Ukraynaca ve diğer dillerde bilgi sağlanmakta, etikette ve kullanım kılavuzunda yer almaktadır.
Bir tıbbi cihazın güvenli kullanımı için gerekli bilgiler doğrudan cihazın üzerine ve/veya ambalajına veya uygun olan durumlarda taşıma kabına yerleştirilmelidir. Her bir cihazın paketlenmesi mümkün değilse, bilgiler bir veya daha fazla tıbbi cihazla birlikte verilen bir eke yerleştirilmelidir.
Kullanım talimatları tıbbi ürünün ambalajında yer almaktadır.
46. Tıbbi bir cihazın kullanımına ilişkin bilgiler semboller şeklinde sağlanabilir. Tüm semboller veya tanımlama renkleri ilgili standartların gerekliliklerine uygun olmalıdır. Bu standartların bulunmadığı sektörlerde, sembol ve renklerin açıklaması ürüne eşlik eden belgelerde yer almalıdır.
47. Tıbbi cihazın etiketinde şunlar belirtilmelidir:
1) Üreticinin adı ve konumu. Piyasaya arz edilmek üzere ithal edilen tıbbi cihazlar için, üreticinin Ukrayna’da kayıtlı bir temsilci ofisi yoksa, etiket, dış ambalaj veya kullanım kılavuzunda, üreticinin yetkili temsilcisinin veya Ukrayna’da kayıtlı sorumlu tedarikçinin adı ve konumu yer almalıdır;
2) Kullanıcının tıbbi cihazı ve yapılandırmasını tanımlaması için gerekli veriler;
3) “STERİL” kelimesi, “BATCH” kelimesinden sonraki parti kodu veya seri numarası, ürünün güvenli kullanımının garanti edildiği tarih; tek kullanımlık tanımlamaya ilişkin bilgi (gerekirse);
4) “özel yapım ürün” (tıbbi cihaz özel yapımdır) ve “sadece klinik araştırma amaçlıdır” (klinik araştırma amaçlıdır) ifadeleri;
5) Tıbbi cihazın saklanması ve/veya kullanımı için özel koşullar, özel kullanım talimatları ve önlemler ve/veya uyarılar;
6) Aktif tıbbi cihazlar için üretim yılı;
7) Steril tıbbi cihazlar için sterilizasyon yöntemi.
48. Tıbbi cihazın kullanım amacı kullanıcı tarafından açıkça anlaşılamıyorsa, üretici bunu etikette ve kullanım kılavuzunda açıkça belirtmelidir.
Tıbbi cihazlar ve yedek parçaları, bu tıbbi cihazlar ve yedek parçalarıyla ilgili olası risklerin tespit edilebilmesi için parti numaralarıyla tanımlanmalıdır.
49. Gerektiğinde kullanım talimatlarında şunlar belirtilmelidir:
1) Bu Teknik Yönetmeliğin 47 nci maddesinde (3 ve 4 üncü alt maddeler hariç) belirtilen bilgiler;
2) performans özellikleri ve herhangi bir istenmeyen yan etki;
3) bu ürün veya ekipmanın özelliklerinin, doğru seçimi ve güvenli bir şekilde birlikte kullanımı için yeterli olacak şekilde ayrıntılı bir açıklaması (tıbbi cihazın amaçlandığı şekilde çalışması için diğer tıbbi ürün veya ekipmanlara monte edilmesi veya bağlanması gerekiyorsa);
4) Tıbbi cihazın doğru şekilde kurulduğunu ve güvenli şekilde kullanıldığını doğrulamak için gerekli olan bilgilerin tamamı, ayrıca cihazın kullanım ömrü boyunca doğru ve güvenli bir şekilde çalışmasını sağlamak için bakım ve kalibrasyonun niteliği ve sıklığı hakkında bilgiler;
5) Tıbbi cihazın yerleştirilmesiyle ilişkili risklerden kaçınmak için gerekli bilgiler;
6) Özel muayene veya tedavi sırasında tıbbi cihazın bulunduğu yerle ilişkili karşılıklı etkileşim riskine ilişkin bilgiler;
7) Steril ambalajın hasar görmesi durumunda gerekli talimatlar ve yeniden sterilizasyon yöntemlerine ilişkin talimatlar;
8) Temizlik, dezenfeksiyon, paketleme, sterilizasyon yöntemi ve yeniden kullanım sayısına ilişkin kısıtlamalar (tıbbi cihazların yeniden kullanılması amaçlanıyorsa) dahil olmak üzere yeniden kullanıma hazırlama işlemlerine ilişkin bilgiler.
Tıbbi cihazların kullanımdan önce sterilizasyona tabi tutulması gerekiyorsa, bu husus ambalaj üzerinde veya cihazın üzerinde belirtilir. Temizlik ve sterilizasyon talimatları, bu Teknik Yönetmeliğin 19-24 üncü maddelerinde belirtilen gerekliliklere uygun olmalıdır;
9) Tıbbi cihazın kullanılmasından önce gerçekleştirilmesi gereken işlemlerin ayrıntıları (örneğin sterilizasyon, son montaj);
10) Bu radyasyonun niteliği, türü, yoğunluğu ve dağılımı hakkında ayrıntılı bilgi (tıbbi cihazlar tıbbi amaçlı radyasyon amaçlı ise).
Tıbbi cihazların kullanım talimatlarında ayrıca, tıbbi personelin hastayı tüm kontrendikasyonlar ve ayrıca aşağıdakiler hakkında uyarmasını sağlayacak bilgiler de yer almalıdır:
Tıbbi cihazların performans özelliklerinde değişiklik olması durumunda alınacak önlemler;
Manyetik alanlara, dış elektriksel dalgalanmalara, elektrostatik deşarja, basınca veya basınç dalgalanmalarına, ivmelenmeye, termal ateşleme kaynaklarına vb. maruz kalmaya karşı önlemler;
tıbbi ürünler veya tıbbi cihazlarla birlikte kullanılmak üzere tasarlanmış ürünler, bu ürünlerin seçimine ilişkin her türlü kısıtlama dahil;
Tıbbi cihazların imhasıyla ilişkili özel, olağandışı tehlikelere karşı önlemler;
tıbbi cihazların ayrılmaz bir parçası olan ilaçlar;
Ölçme fonksiyonuna sahip tıbbi cihazlar için belirlenen doğruluk derecesi.
50. Bu Teknik Yönetmelikte belirlenen gerekliliklere uyumun klinik verilere dayanması halinde, bu veriler DSTU 4659-1-2:2006 “İnsan Kullanımına Yönelik Tıbbi Cihazların Klinik Araştırmaları” (ISO 14155-1-2; 2003, MOD) uyarınca tespit edilir.
Genel gereksinimler
51. Üretici şunları yapmalıdır:
1) Tıbbi cihazların işleyişinin sistematik analizini yapmak ve tıbbi cihazın özelliklerini ve/veya niteliklerini ve bununla ilişkili riskleri dikkate alarak gerekli düzeltici eylemleri almak için uygun önlemleri almak;
2) Aşağıdaki durumlarda derhal Sağlık Bakanlığı’na haber vermek:
Tıbbi cihazın özelliklerinde ve/veya performansında meydana gelen herhangi bir arıza veya bozulma, ayrıca etiket veya kullanım kılavuzunda hastanın, kullanıcının ve diğer kişilerin ölümüne veya sağlığında önemli bozulmaya yol açabilecek herhangi bir tutarsızlık;
Bu fıkranın 1. alt fıkrasında belirtilen nedenlerden dolayı, tıbbi cihazın özelliklerine ve/veya performansına ilişkin olarak üreticinin aynı tipteki tıbbi cihazları piyasadan çekmesine yol açan her türlü teknik veya tıbbi çekince.
Uygunluk değerlendirme prosedürleri
52. Ulusal uygunluk işaretini taşıyan tıbbi cihazların uygunluk değerlendirmesini gerçekleştirmek için, 7 Ekim 2003 tarihli ve 1585 sayılı “Uygunluk Değerlendirme Modüllerinin Teknik Yönetmeliklerinin Onaylanması ve Teknik Yönetmeliklerde Uygulanan Ulusal Uygunluk İşareti ile İşaretleme Gereksinimleri Hakkında” (Ukrayna Resmi Gazetesi, 2003, No. 41, s. 2175; 2007, No. 1, s. 31) Ukrayna Bakanlar Kurulu Kararı uyarınca uygunluk değerlendirme prosedürleri modülleri veya bunların kombinasyonları uygulanmalıdır. Bu kararda, tıbbi cihazların kullanımının aşağıdaki özellikleri dikkate alınmalıdır:
1) Modül A (dahili üretim kontrolü) için:
İç üretim kontrolü, bu Teknik Yönetmeliğin 19-50 nci maddelerinin gereklerine ve steril ürünler ile ölçüm fonksiyonlu ürünlerin piyasaya arzı durumunda bu Teknik Yönetmeliğin bu maddesinin gereklerine uyan üretici veya yetkili temsilcisinin, tıbbi cihazların belirlenen gereklere uygunluğunu garanti ettiği ve beyan ettiği bir uygunluk değerlendirme prosedürüdür;
Üretici veya yetkili temsilcisi, her tıbbi cihaza ulusal uygunluk işaretini iliştirir ve ekte belirtilen biçimde bir uygunluk beyanı düzenler;
teknik dokümantasyon şunları içermelidir:
– Sterilizasyon yöntemlerinin açıklaması (tıbbi cihazlar steril halde piyasaya arz ediliyorsa);
– Üretici tarafından belirtilen özelliklere sahip bir ürüne bağlandığında gereklilikleri karşıladığının teyidi (tıbbi cihazın amaçlanan kullanımı için başka bir tıbbi cihaza/cihazlara bağlanması durumunda);
– etiketleme ve kullanım talimatları.
Steril halde piyasaya arz edilen tıbbi cihazlar ve ölçüm fonksiyonlu sınıf I cihazlar için üretici, ayrıca modül D veya modül E veya modül F’ye uygun uygunluk değerlendirme prosedürlerinden birini gerçekleştirmelidir.
Bu prosedürlerin uygulanması aşağıdakilerle sınırlıdır:
Steril olarak piyasaya arz edilen tıbbi cihazlar için, yalnızca sterilitenin sağlanması ve sürdürülmesine ilişkin üretim süreci ile;
Ölçüm fonksiyonu olan ürünler için – yalnızca ürünlerin metrolojik gerekliliklere uygunluğu ile ilgili üretim süreci.
Bu tür bir modülün, modül D veya modül E veya modül F’de öngörülen prosedürlerle birlikte sınıf IIa ürünlerine uygulanması durumunda, üretici, bu Teknik Yönetmeliğin bu tür bir modüle uygulanabilir gerekliliklerine uygunluğunu teyit ve beyan ettiği tek bir beyanname düzenler;
2) B modülü (tip incelemesi) için – teknik dokümantasyon şunları içermelidir:
tasarım hesaplamaları, risk analizi, gerçekleştirilen araştırma ve teknik testlerin sonuçları;
Ürünün, bu Teknik Yönetmeliğin 4 üncü maddesine uygun bir maddeyi ayrılmaz bir parça olarak içerip içermediğini ve test sonuçlarını belirten bir bildirim;
taslak etiket ve gerekirse kullanım talimatları.
Tıbbi cihazlar, bu Teknik Yönetmeliğin 4 üncü maddesinde belirtilen bir maddeyi ayrılmaz bir parça olarak içeriyorsa, yetkili kuruluş, bu fıkra hükümlerini dikkate alarak, karar vermeden önce söz konusu maddenin kullanımı konusunda Sağlık Bakanlığından görüş veya açıklama almak zorundadır;
3) Modül D (üretim kalite güvencesi) için:
Tıbbi cihazlar için kalite yönetim sisteminin, DSTU ISO 13485:2005 “Tıbbi cihazlar. Kalite yönetim sistemi. Mevzuat gereklilikleri (ISO 13485:2003, IDT)” standardının 7.3 maddesi hariç, işletmede uygulanması halinde bu gereklilikleri karşıladığı kabul edilir;
Tıbbi cihazların kalite yönetim sisteminin değerlendirilmesi için üretici, kendi seçtiği yetkili kuruluşa aşağıdakileri içeren bir başvuruda bulunmalıdır:
– üreticinin adı ve yeri;
– Kalite yönetim sistemi kullanılarak üretilen bir tıbbi cihaza ilişkin bilgiler;
– böyle bir başvurunun başka bir yetkili makama sunulmadığına dair yazılı onay;
– kalite yönetim sistemine ilişkin dokümantasyon;
– Gerektiğinde, test edilen tipe ait teknik dokümanlar ve tip inceleme sertifikasının bir kopyası;
– Tıbbi cihazların işleyişinin sistematik analizine ilişkin bilgilerin uygun önlemlerin alınması için kullanılması.
Bu tür bir modülün IIa sınıfı ürünlere uygulanması durumunda, üretici, söz konusu ürünlerin A modülüne uygun olarak düzenlenen teknik belgelere uygun olduğunu ve bu Teknik Yönetmeliğin kendilerine uygulanan gereklerine uyulmasını garanti ve beyan ettiği bir uygunluk beyanı düzenler;
4) E modülü (ürün kalite güvencesi) için – steril halde piyasaya arz edilen tıbbi cihazlar için, bunu sağlamak ve sürdürmek amacıyla üretici, D modülüne uygun olarak onaylanmış kuruluşun kalite yönetim sistemi ve gözetimi için belirtilen hükümleri uygulamalıdır.
Yetkili kuruluş, kalite yönetim sisteminin DSTU ISO 13485:2005 “Tıbbi cihazlar. Kalite yönetim sistemi. Mevzuat gereklilikleri (ISO 13485:2003, IDT)” gerekliliklerine uygunluğunu değerlendirir.
Bu modülün IIa sınıfı ürünlere uygulanması durumunda, üretici, söz konusu ürünlerin A modülüne göre düzenlenen teknik dokümanlara uygunluğunu garanti ve beyan ettiği ve bu Teknik Yönetmelik hükümlerine uyulmasını sağladığı bir uygunluk beyanı düzenler;
5) F modülü (ürün doğrulaması) için – tıbbi cihazın imalatına başlamadan önce üretici, özellikle sterilizasyon olmak üzere üretim sürecine ilişkin dokümantasyonu, üretimin tekdüzeliğini ve ürünün tip inceleme sertifikasında belirtilen tipe uygunluğunu garanti eden hükümleri ve bu Teknik Yönetmeliğin kendisine uygulanan gerekliliklerini dikkate alarak hazırlamalıdır.
Ayrıca, steril halde piyasaya arz edilen tıbbi cihazlar için, bu durumu sağlamak ve sürdürmek amacıyla üretici, Modül D uyarınca onaylanmış kuruluşun kalite yönetim sistemi ve gözetimine ilişkin olarak belirlenen hükümleri uygulamalıdır.
Bu tür bir modülün IIa sınıfı ürünlere uygulanması durumunda, üretici, söz konusu ürünün A modülüne uygun olarak düzenlenen teknik dokümantasyona uygun olduğunu ve bu Teknik Yönetmeliğin kendilerine uygulanan gerekliliklerine uyulmasını garanti ve beyan ettiği bir uygunluk beyanı düzenler;
6) H modülü (tam kalite güvencesi) için – onaylanmış kuruluş, işletmedeki kalite yönetim sisteminin DSTU ISO 13485:2005 “Tıbbi cihazlar. Kalite yönetim sistemi. Mevzuat gereklilikleri (ISO 13485:2003, IDT)” gerekliliklerine uygunluğunu belirlemek için değerlendirme yapar.
Bu modül, tasarım incelemesine ilişkin hükümler olmaksızın IIa ve IIb sınıfı ürünlere uygulanabilir.
Farklı tıbbi cihaz sınıflarının uygunluk değerlendirmesi
53. Sınıf III olarak sınıflandırılan, özel olarak üretilmemiş veya klinik araştırmalar için tasarlanmamış tıbbi cihazlar, üreticinin tercihine bağlı olarak modül H veya modül B ile modül D veya modül F kombinasyonuna göre uygunluk değerlendirmesine tabi tutulur.
Sınıf IIa olarak sınıflandırılan, özel olarak üretilmeyen veya klinik araştırmalar için tasarlanmamış tıbbi cihazlar, üreticinin tercihine bağlı olarak, tasarım incelemesi olmaksızın modül H’ye veya modül D veya modül F veya modül E ile birlikte modül A’ya göre uygunluk değerlendirmesine tabi tutulur.
Sınıf IIb olarak sınıflandırılan, özel olarak üretilmeyen veya klinik araştırmalar için tasarlanmamış tıbbi cihazlar, üreticinin tercihine bağlı olarak, tasarım incelemesi olmaksızın modül H’ye veya modül D veya modül F veya modül E ile birlikte modül B’ye göre uygunluk değerlendirmesine tabi tutulur.
Sınıf I olarak sınıflandırılan, özel olarak üretilmeyen veya klinik araştırmalar için tasarlanmamış tıbbi cihazlar, Modül A’ya göre uygunluk değerlendirmesine tabidir.
Özel yapım tıbbi cihazların dolaşıma girmesi ve çalıştırılması
54. Özel olarak üretilen tıbbi cihazların piyasaya arzı ve işletmeye alınması için imalatçı veya yetkili temsilcisi, aşağıdaki hususları belirten bir bildirim düzenlemek zorundadır:
ilgili ürünü tanımlamak için gerekli bilgiler;
Bu ürünün yalnızca belirli bir hasta tarafından kullanılmak üzere tasarlandığına dair bilgi, bu hastanın soyadı;
Bu randevuyu veren doktorun veya yetkili kişinin adı, kliniğin adı;
Ürünün tıbbi amacına göre özel nitelikleri;
Tıbbi cihazın bu Teknik Yönetmelikte belirtilen güvenlik şartlarına uygunluğu ve bu Teknik Yönetmelik şartlarının tam olarak karşılanmaması halinde uygun gerekçe.
Özel olarak üretilen tıbbi cihazlara ulusal uygunluk işareti uygulanmaz.
Ayrıca üretici, ürünün tasarımını, beklenenler de dahil olmak üzere performans özelliklerini anlamayı ve bu Teknik Yönetmelik hükümlerine uygunluğu tespit etmeyi mümkün kılan belgeleri düzenlemek ve beş yıl süreyle saklamak zorundadır.
Üretici, üretim sürecinin düzenlenen belgelere uygun olmasını sağlayacak tedbirleri almak zorundadır.
Özel üretim tıbbi cihaz üreticilerinin, bunların listesini Sağlık Bakanlığı’na sunmaları gerekiyor.
Üretici ve/veya onaylanmış kuruluş, bir tıbbi cihaza ilişkin uygunluk değerlendirme işlemini gerçekleştirirken, söz konusu değerlendirmenin sonuçlarını ve üretimin ara aşamasında bu Teknik Yönetmelik uyarınca gerçekleştirilen denetimleri dikkate almak zorundadır.
Üretici, tıbbi cihazın kullanımına ilişkin potansiyel risklere bağlı olarak, onaylı modüllere uygun olarak uygunluk değerlendirme onay prosedürlerinin uygulanması konusunda yetkili temsilcisine talimat verme hakkına sahiptir.
İstisnai olarak, Sağlık Bakanlığı, talep üzerine, belirtilen işlemleri yapılmamış ancak kamu sağlığının korunması açısından kullanımı zorunlu olan bireysel tıbbi cihazların dolaşıma sokulmasına ve çalıştırılmasına izin verebilir.
Sistem ve prosedür komplekslerinin dolaşıma girmesi ve çalıştırılmasının özellikleri
55. Ulusal uygunluk işaretini taşıyan tıbbi cihazları, amaçlanan kullanım kapsamı ve üretici tarafından sistem veya işlem kompleksi olarak piyasaya arz edilmek üzere tanımlanan uygulama kapsamı çerçevesinde bir araya getiren üreticinin yetkili temsilcisi, aşağıdakileri teyit eden bir beyanname düzenlemelidir:
ürün uyumluluğu kontrol edildi, işlemler üreticinin talimatlarına göre gerçekleştirildi;
sistem veya prosedür kompleksini paketledi ve ürünü kullanıcı bilgileri ve üreticinin talimatlarıyla birlikte sağladı;
İç kontrol ve doğrulama yöntemlerine uygun olarak faaliyetlerini yürütür.
Belirtilen şartlar sağlanmıyorsa veya sistem veya prosedür kompleksi ulusal uygunluk işaretini taşımayan tıbbi cihazları içeriyorsa veya cihazların birleşimi amaçlanan amaçları açısından uyumsuzsa, bu sistem veya prosedür kompleksi, uygunluk değerlendirme prosedürünün bu Teknik Yönetmeliğin 53 üncü maddesine uygun olarak gerçekleştirilmesi gereken bağımsız bir tıbbi cihaz olarak kabul edilmelidir.
Üreticinin yetkili temsilcisi, üretici tarafından sağlanan ulusal uygunluk işaretini taşıyan bir sistemi veya işlem setini veya diğer tıbbi cihazı kullanımdan önce sterilize etmişse, kendi tercihine göre modül F, modül D veya modül E’deki işlemlerden birini gerçekleştirmelidir.
Bu Teknik Yönetmeliğin 54-56 ncı maddelerinde belirtilen tıbbi cihazlar, ek bir ulusal uygunluk işareti taşımaz. Bu tür cihazlara, bu Teknik Yönetmeliğin 45-50 nci maddelerinde belirtilen bilgiler eşlik eder.
Belirtilen ürünlere ilişkin beyanların en az beş yıl süreyle saklanması zorunludur.
Klinik araştırmalara yönelik tıbbi cihazların dolaşıma sokulması ve çalıştırılmasının özellikleri
56. Klinik araştırmalara yönelik tıbbi cihazların piyasaya arzı ve işletmeye alınması için imalatçı veya yetkili temsilcisi;
ürün tanımlaması için gerekli bilgiler;
amacını, bilimsel veya tıbbi gerekçelerini, planlanan çalışmanın kapsamını ve incelenecek tıbbi cihaz sayısını tanımlayan bir araştırma programı;
Etik kurul kararı ve benzer çalışmaların sonuçları;
Klinik araştırmaları yürütmeye yetkili hekimin veya diğer kişinin adı, araştırmaları yürütmeye yetkili kurumun adı;
araştırmanın yeri, tarihi ve süresi.
Üretici aşağıdaki belgeleri hazırlayacak ve en az beş yıl süreyle saklayacaktır:
ürünün genel tanımı;
tasarım çizimleri, sterilizasyon dahil üretim yöntemleri ve bileşenlerin ve montajların diyagramları;
Belirtilen çizimlerin, şemaların ve ürünün çalışma prensibinin anlaşılması için gerekli açıklamalar ve tanımlamalar;
Tıbbi cihazın kullanımına ilişkin risk analizinin sonuçları, ulusal standartların tam veya kısmi olarak uygulanması dikkate alınarak, standartlar tam olarak uygulanmadığı takdirde bu Teknik Yönetmelik gereklerini karşılamak için alınan kararların açıklamaları;
tasarım hesaplamaları ve gerçekleştirilen metrolojik kontroller ve teknik testlerin sonuçları.
Üretici, tıbbi cihazların belirtilen dokümanlara uygun olarak üretilmesini sağlamak için gerekli tedbirleri almalıdır.
Üretici, bu önlemlerin etkinliğini değerlendirmek için bir test yapılmasına izin verir.
Klinik araştırmalara yönelik tıbbi cihazlara ulusal uygunluk işareti yapıştırılmaz.
, DSTU 4659-1-2:2006 “İnsanlar için tıbbi cihazların klinik araştırmaları” standardına uygun olarak tıbbi cihazların klinik araştırmalarının yürütüldüğünü Sağlık Bakanlığına bildirir.
Bu maddenin hükümleri, bu Teknik Yönetmelik hükümlerine uygun olarak ulusal uygunluk işareti iliştirilmiş tıbbi cihazlar kullanılarak yürütülen klinik araştırmalara uygulanmaz; ancak bu cihazların uygunluk değerlendirme prosedüründe belirtilen amaçlar dışında kullanılmaması gerekir. Bu tür klinik araştırmalar, DSTU 2459-1-2:2006 “İnsanlar için Tıbbi Cihazların Klinik Araştırmaları” standardına uygun olarak yürütülür.
______________________



